One of the longest-standing issues in the development of advanced molecular materials for applications in technology concerns the following question: how does the molecular structure affects the properties of the “aggregated” materials? Generally speaking, this question is at the basis of crystal structure prediction (CSP). The relationship between molecular structure and, for example, solid-state packing, is extremely relevant in the development of new drugs. However, the same question arises in the development of applications based on molecular materials. In many applications, organic molecules are deposited onto a substrate to form a thin-film at the interface. The properties of the molecular materials at the interface, in turn, may control several critical processes in applications.[1][2]
The properties of molecular aggregates depend crucially on the peculiar molecular structure.[3] This means that even tiny changes of the molecular structure can alter significantly the local and long-range aggregation, leading to potentially strong modifications of the materials properties.[4] However, the formation of molecular aggregates onto substrates constitutes a subset of the incredible task of determining the many likely ways a molecule can aggregate with other molecules of the same kind. Molecular aggregation on surfaces is ruled essentially by two driving forces: 1. The molecule-molecule interaction and 2. The molecule-substrate interaction. The interplay between these two components, also balanced by the external conditions, determines the growth of molecular aggregates onto substrates. These interactions are governed by weak (van der Waals) forces, leading to a very complex landscape, featuring several different aggregation morphologies in which the system can likely be trapped.
Unfortunately, generic CSP approaches are not yet successful in predicting the morphology of molecular aggregates as a function of the molecular structure and in different conditions and environments. The majority of predictive approaches relies on the evaluation and ranking of different aggregation morphologies. In principle, the particular case of growth of molecular aggregates on substrate could be simulated by molecular dynamics (MD), throwing individual molecules towards a bare substrate, one after the other, until the desired surface density is reached. This “direct” method clashes with one of the major limitations of atomistic MD methods, that is, relatively short simulation timescales. For nanosized systems, MD can reach a few microseconds of simulation time, which is, unfortunately, several orders of magnitudes shorter than real deposition times. For example, if we simulate by MD the growth onto a flat substrate of a molecular monolayer with a height of 1 nm in 1us, we have a growth rate of about 1e6 nm/second, which is about 1e6 times (!) larger than a realistic rate (about 0.1-0.01 Ang/min). In addition, the simulation itself could take several days even on a supercomputer. This means that, in the best case, with “direct” MD we are simulating a particular growth process, in which we do not provide the molecules with enough time to “relax” energetically. Of course, this is a typical issue related to all MD simulations (just to mention one example, protein folding is also difficult to simulate by MD for similar reasons). However, to simulate aggregation onto a surface we can take advantage of the specific conditions and think about some hack.
We can apply two principles:
- Simulation temperature accelerates the dynamics of molecular systems. In our simulated world, we can assign different temperatures to different parts of a complex system, with the purpose of triggering different dynamical effects.
- The relative strength of the molecule-molecule interaction, with respect to the molecule-substrate interaction, can be tweaked in simulations. This would change the energy landscape that controls aggregation.
We can use these two “hacks” for the definition of a relatively simple computational protocol for the simulation of the growth of molecular aggregates onto substrates. The simulation steps required are shown below:
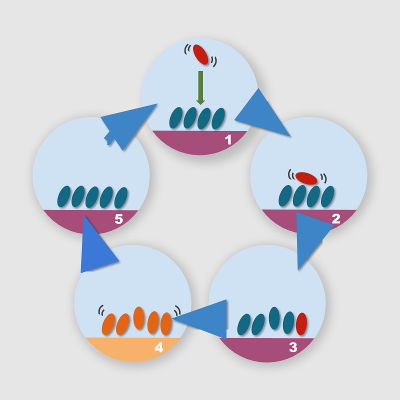
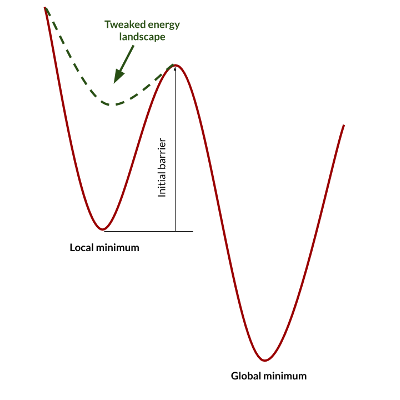
With these principles in mind, we can play with the simulation conditions and interaction potential to favor some processes with respect to others, and consequently observe different aggregation morphologies. The modification of interaction potentials and the differential equilibrium temperatures lead to a strong unbalancing of the initial energy landscape, leading to processes that occur within times that are compatible with the typical simulation times of molecular dynamics. If for example we reduce the interaction energy of a system that is trapped into a local minimum, we may lower the barrier for transition to a lower energy minimum. If we subsequently restore the pristine interaction potential, we finally get a thermally equilibrated system in a different state:
We applied this protocol to predict the morphology of a molecular system, a perylene diimide derivative, which is used in organic electronics, onto graphene. In principle, we can extend the same simulation protocol to other molecules and substrates.
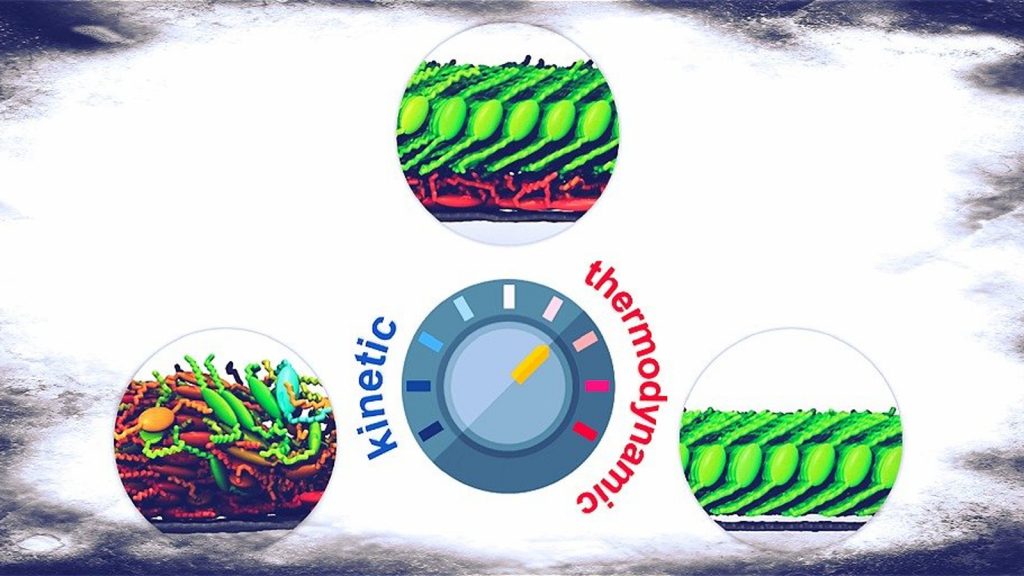
By playing with growth conditions, we obtained a set of different aggregation morphologies, from very disordered to fully ordered aggregates:
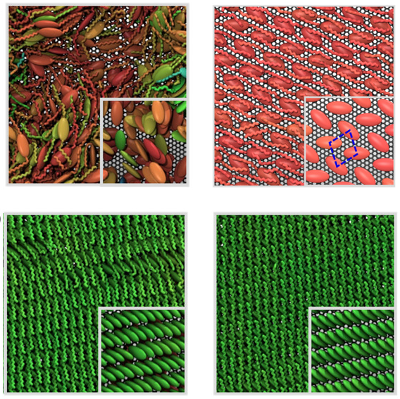
The predicted morphologies are in excellent agreement with the available experiments. The structure of molecular aggregates in kinetically- or thermodynamically-controlled conditions (or even in intermediate conditions) are correctly reproduced. Moreover, simulations allow us to predict several aspects of molecular growth and aggregation, including, for example, the occurrence of grain boundaries.
The simulation protocol is therefore able to link molecular structure, processing conditions and resulting morphology of molecular aggregates at the interface with a substrate. The predictive power of this approach enables a step forward in the engineering of molecular structures for applications in technology.
Part of this work has recently been published on Advanced Theory and Simulations [5].
References
[1] Correlation between gate-dielectric morphology at the nanoscale and charge transport properties in organic field-effect transistors. A Lorenzoni, M Muccini, F Mercuri. RSC Advances 5 (16), 11797-11805, 2015.
[2] Nanoscale morphology and electronic coupling at the interface between indium tin oxide and organic molecular materials. A Lorenzoni, AM Conte, A Pecchia, F Mercuri. Nanoscale 10 (19), 9376-9385, 2018.
[3] Theoretical insights on morphology and charge transport properties of two-dimensional N, N′-ditridecylperylene-3, 4, 9, 10-tetra carboxylic diimide aggregates. A Lorenzoni, F Gallino, M Muccini, F Mercuri. RSC Advances 6 (47), 40724-40730, 2016.
[4] Redox-switchable devices based on functionalized graphene nanoribbons. D Selli, M Baldoni, A Sgamellotti, F Mercuri. Nanoscale 4 (4), 1350-1354, 2012.
[5] A Computational Predictive Approach for Controlling the Morphology of Functional Molecular Aggregates on Substrates. A Lorenzoni, M Muccini, F Mercuri. Adv. Theory Simul. 2, 1900156, 2019.